Submitting Jobs
Browsing Results
- Browse Main page
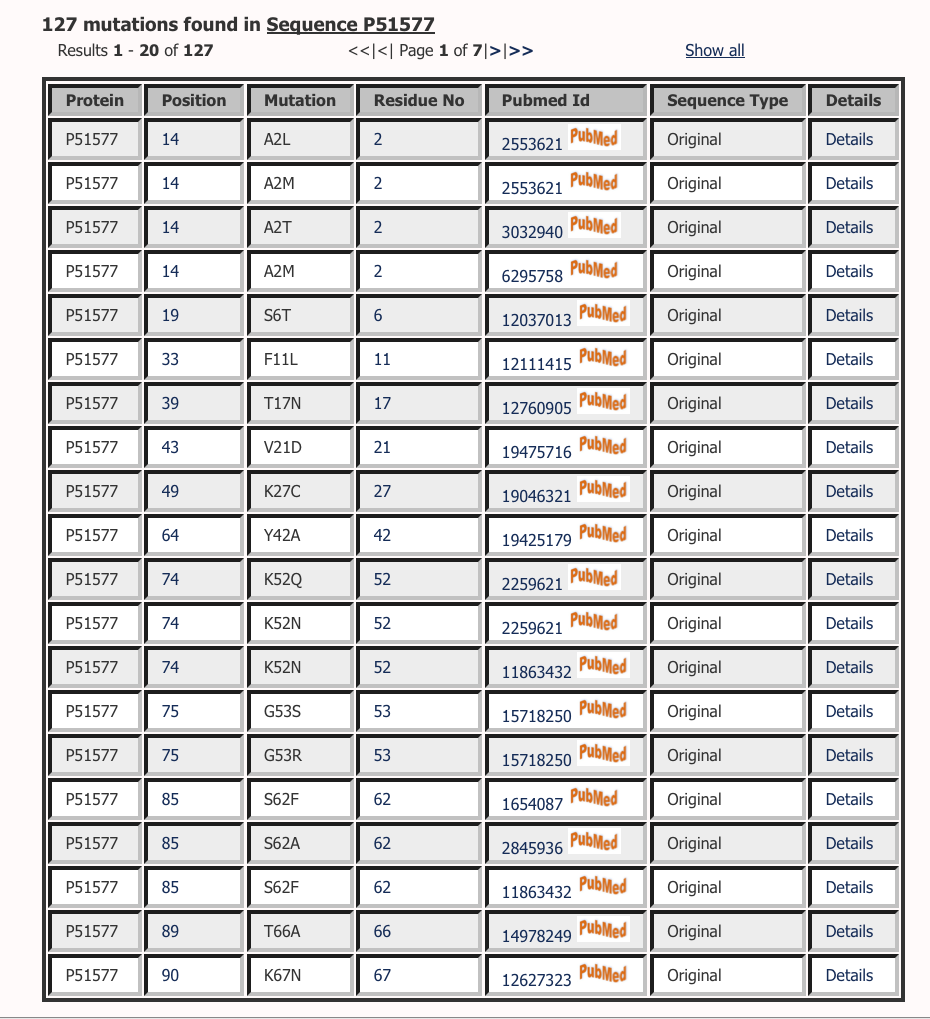
- Browse by Sequence
- Browse by Alignment position
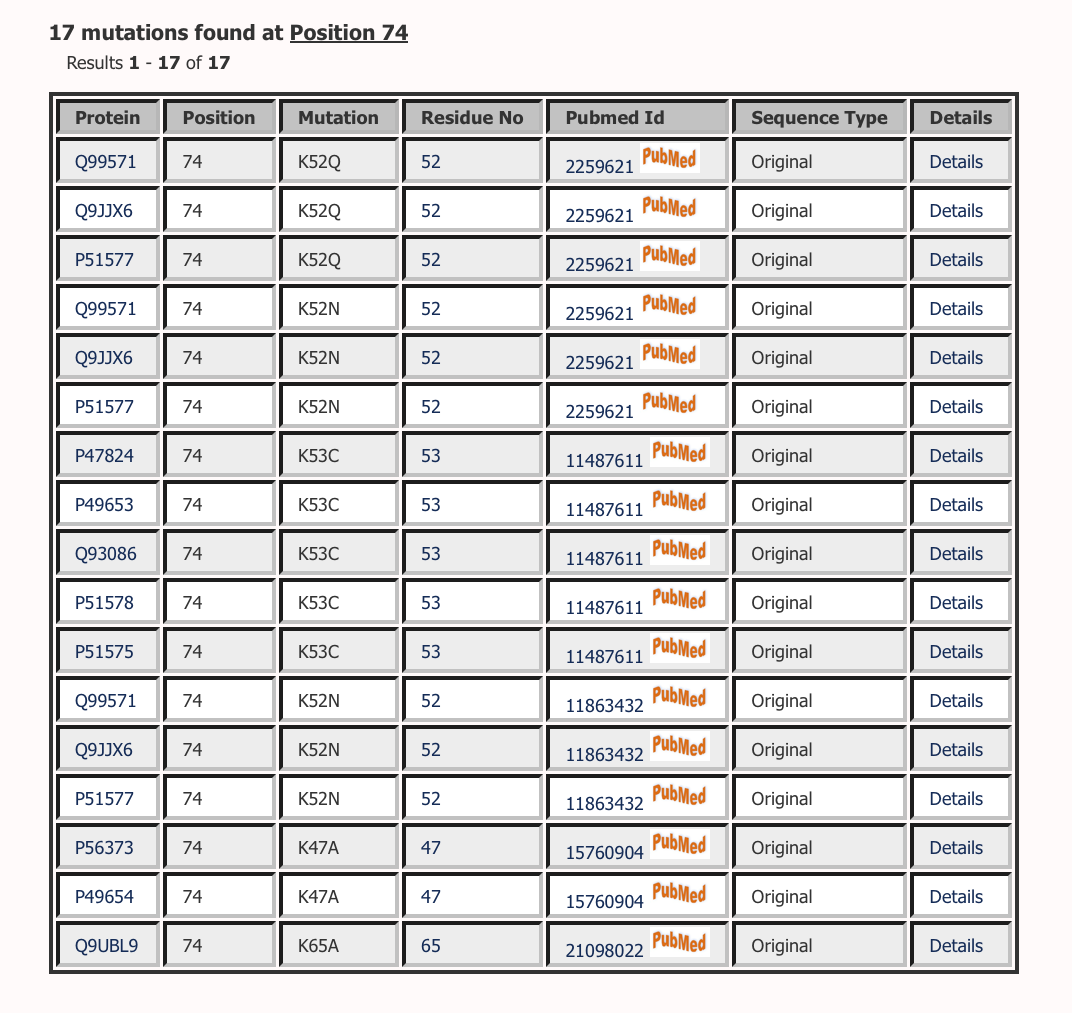
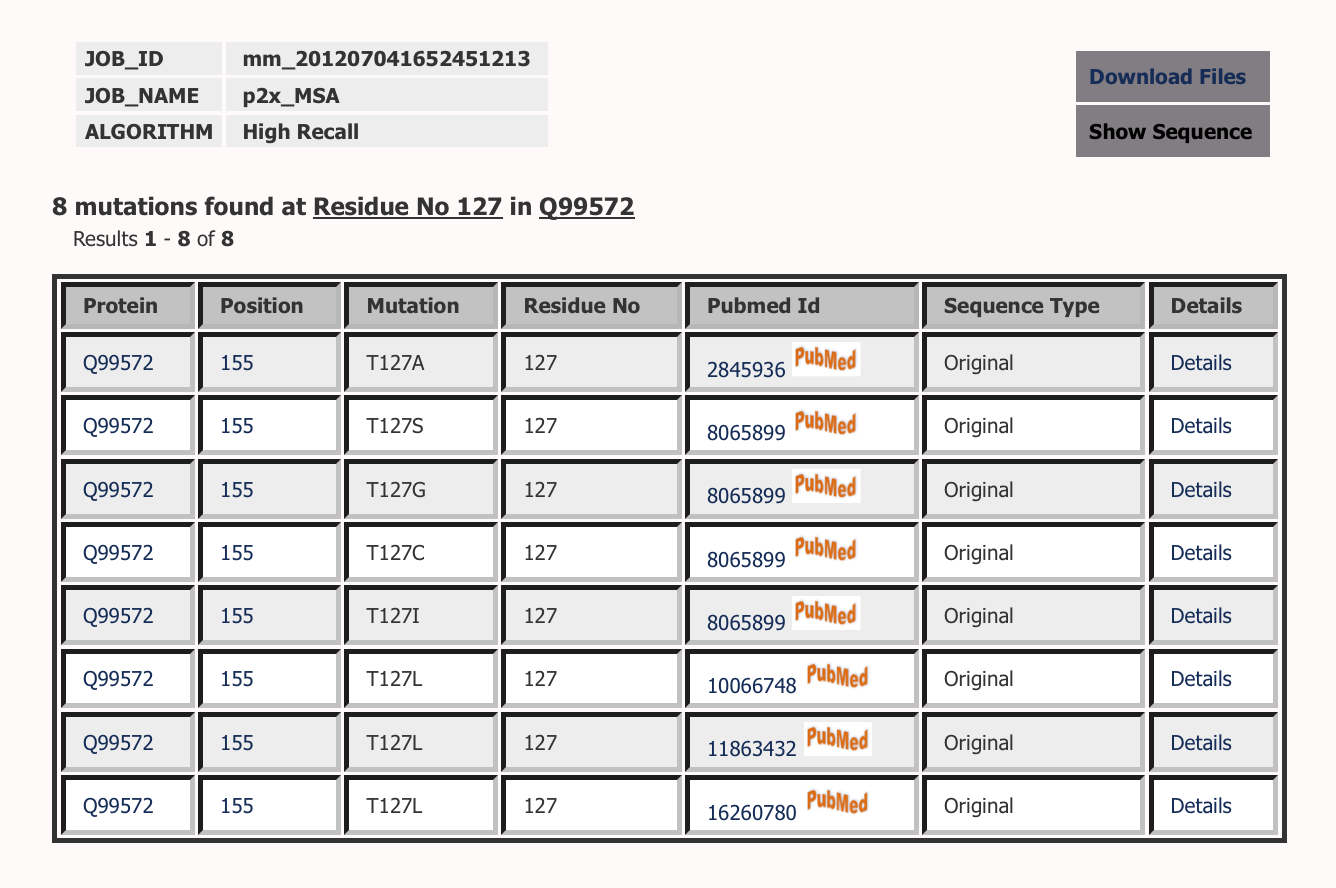
- Browse by Residue number
- Browse by Pubmed Id
- Browse Details
Extract and Map Point Mutations
MutationMapper is a tool to identify, extract and map point mutations from literature on the protein reported. It uses a protein sequence or a multiple sequence alignment in fasta format as input. It uses a Uniprot ID or an Accession code provided as a descriptor to retrieve protein information from Uniprot. From there, it extracts protein names , gene names and synonyms. These are used as keywords to download abstracts. The retrieved abstracts are scanned for mutations. Mutations reported are mapped onto the amino acid sequence.
Input can be a Uniprot Id, single sequence or multiple sequence alignment.
It accepts one uniprot ID at a time and not a list of IDs.
The amino acid sequence should be submitted in FASTA format.
FASTA format consists of a single-line description which should be Uniprot Id or Accession code of the sequence, followed by lines
of amino acid sequence.
The first character of the description line should be a greater-than
(">") symbol:
Q9Z1M0or P2RX7_MOUSE |
>Q9Z1M0 MPACCSWNDVFQYETNKVTRIQSTNYGTVKWVLHMIVFSYISFALVSDKLYQRKEPVISS VHTKVKGIAEVTENVTEGGVTKLGHSIFDTADYTFPLQGNSFFVMTNYVKSEGQVQTLCP EYPRRGAQCSSDRRCKKGWMDPQSKGIQTGRCVPYD |
>Q9Z1M0 -------------------MPACCSWNDVFQYETNKVTRIQSTNYGTVKWVLHMIVFSYI S-FALVSDKLYQRKEP-VISSVHTKVKGIAEVTENVTEGGVTKLGHSIFDTADYTFPLQG -NSFFVMTNYVKSEGQVQTLCPEYP-RRGAQCSSDRRCKKGWMDPQSKGIQTGRCVPYD- >Q64663 -------------------MPACCSWNDVFQYETNKVTRIQSVNYGTIKWILHMTVFSYV S-FALMSDKLYQRKEP-LISSVHTKVKGVAEVTENVTEGGVTKLVHGIFDTADYTLPLQG -NSFFVMTNYLKSEGQEQKLCPEYP-SRGKQCHSDQGCIKGWMDPQSKGIQTGRCIPYD- >Q99572 -------------------MPACCSCSDVFQYETNKVTRIQSMNYGTIKWFFHVIIFSYV C-FALVSDKLYQRKEP-VISSVHTKVKGIAEVKEEIVENGVKKLVHSVFDTADYTFPLQG -NSFFVMTNFLKTEGQEQRLCPEYP-TRRTLCSSDRGCKKGWMDPQSKGIQTGRCVVYE- |
JobName: It would be useful to provide a job name in few words but is not mandatory.
Email: You have to provide a vaild email Id. You will get an email confirmation when you submit the job and when it is finished.
The advanced option of input of expression list is useful if the MutationMapper fails to map relevant mutations for the protein of interest. This is because the protein expression retireved from Uniprot may not include the commonly occuring names in abstracts. The user can provide the expression list for better retrieval.
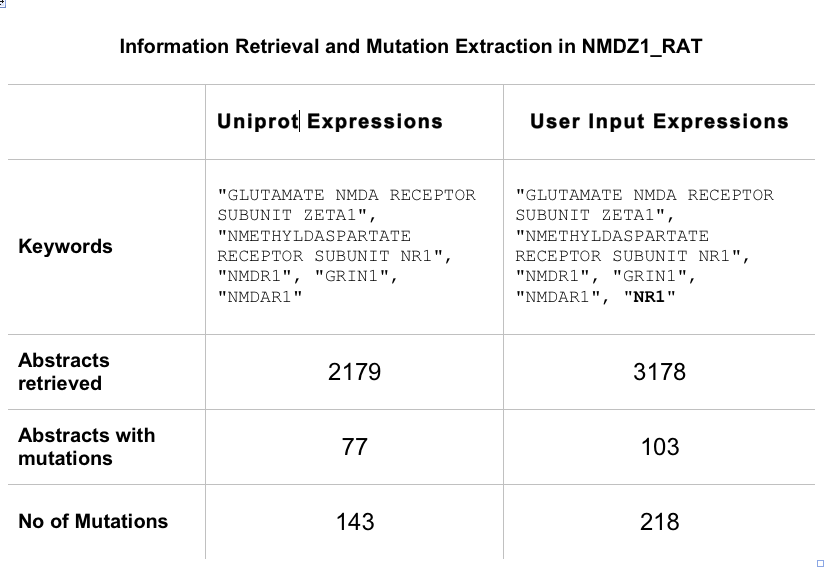
Expression List: It uses Uniprot Identifier followed by list of expressions. The Uniprot identifier should be same as used in the input sequence.
NMDZ1_RAT:GLUTAMATE NMDA RECEPTOR SUBUNIT ZETA1;NMETHYLDASPARTATE RECEPTOR SUBUNIT NR1;NMDR1;GRIN1;NMDAR1; |
where NMDZ1_RAT is the Uniprot Identifier, followed by list of expressions.
NMDE1_RAT:GLUTAMATE NMDA RECEPTOR SUBUNIT EPSILON1;NMETHYL DASPARTATE RECEPTOR SUBTYPE 2A;NMDAR2A;NR2A;GRIN2A; NMDE2_RAT:GLUTAMATE NMDA RECEPTOR SUBUNIT EPSILON2;NMETHYL DASPARTATE RECEPTOR SUBTYPE 2B;NMDAR2B;NR2B;GRIN2B; NMDE3_RAT:GLUTAMATE NMDA RECEPTOR SUBUNIT EPSILON3;NMETHYL DASPARTATE RECEPTOR SUBTYPE 2C;NMDAR2C;NR2C;GRIN2C; NMDE4_RAT:GLUTAMATE NMDA RECEPTOR SUBUNIT EPSILON4;NMETHYL DASPARTATE RECEPTOR SUBTYPE 2D;NMDAR2D;NR2D;GRIN2D; |
On submission, you will be given a JobId which can be used to browse the status of the job. If the server is busy, your job will be in a queue. The status would be updated once the job starts running. You can view the status by entering the JobId on the status page. You will receive an email once the job is finished. It may take a while to get the results if the number of abstracts downloaded to scan for mutations is far too many.
You can browse the results by clicking on the direct link in your email or entering the JobId on the status page. The main page shows the sequence or the alignment and highlighted residues are mutations mapped.
You can browse the results based on sequence, alignment position, residue number and Pubmed Id.
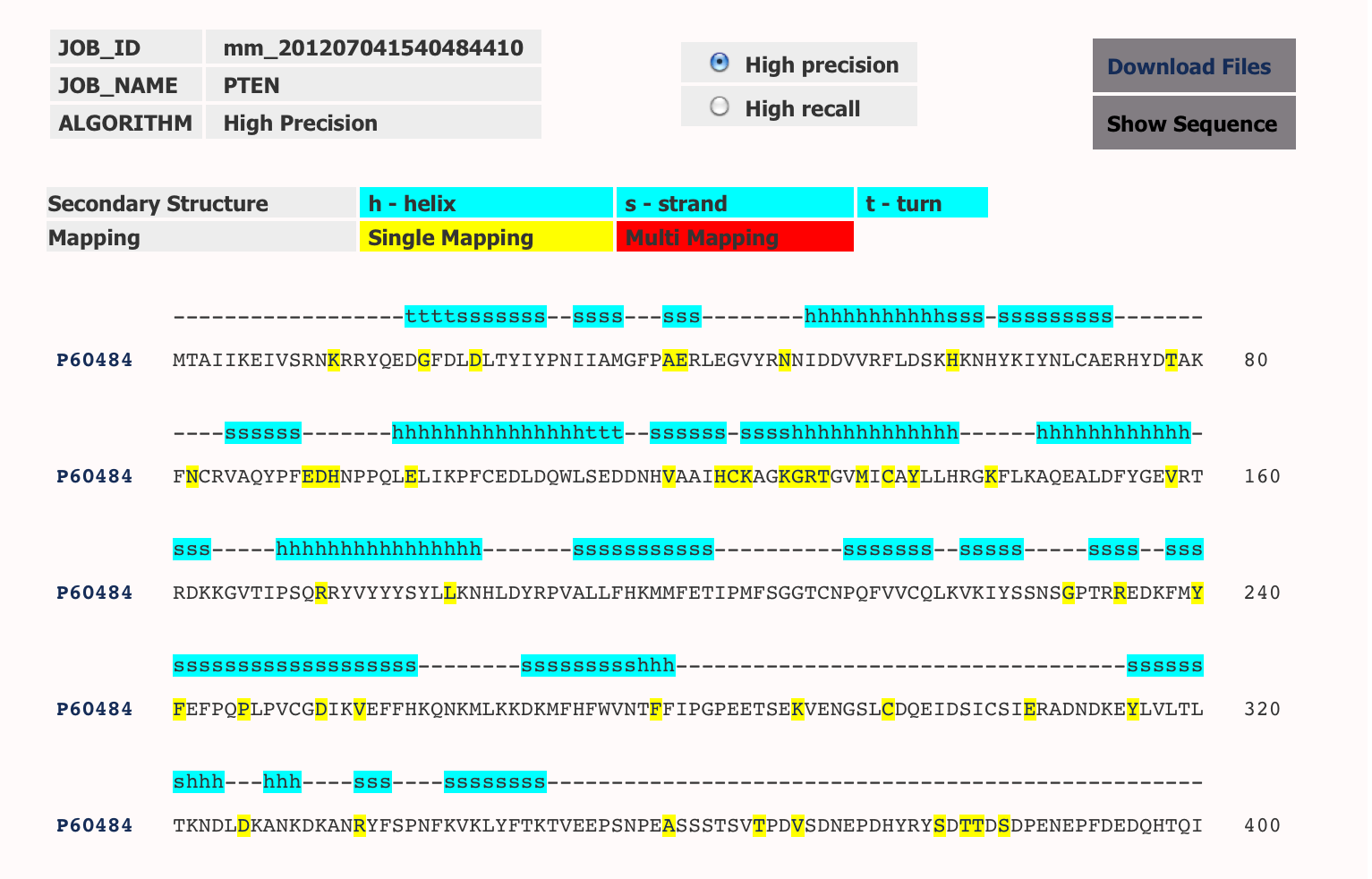
Displays JobId created when you submit the job, jobname provided and the algorithm. More details about algorithms is given below. Default is High precision. You can browse results with High Recall by clicking on the radio button. Mutated residues are highlighted. Residues in yellow are the mutations mapped only on that residue but if the same mutation has been mapped on more than one residue then the residue is coloured red.Secondary Structure has been highlighted in cyan. Secondary structure information has been extracted from Uniprot. We aim to let user input this information in later release. On clicking the residues, it will show all the mutations that have been reported on the position in that particular sequence. On clicking the protein name, it will display all the mutations reported on the protein sequence.
Results below are the mutations mapped on PTEN_HUMAN Sequence.
Results displayed below are the mutations mapped on an alignment of closely related P2X receptor sequences.

Lists all the mutations reported on a protein sequence. You can click on any alignment position to browse all the mutations at that position in all the sequences. Similiarly you can click on the residue position to browse all the mutations reported at that residue position in that particular sequence. Browsing on Pubmed Id will list all the mutations reported in the abstract. Pubmed ID are directly linked to the abstracts. Sequence type defines if the mutation is mapped on original sequence or matured sequence. In many cases, where there is a signal peptide, mutations are mapped on a mature sequence. You can click on details to get more information about the mutation.
Lists all the mutations at a particular position in all protein sequences in Multiple Sequence Alignment.
Lists all the mutations at a particular position in a particular sequence.
Lists all the mutations reported in a particular abstract.
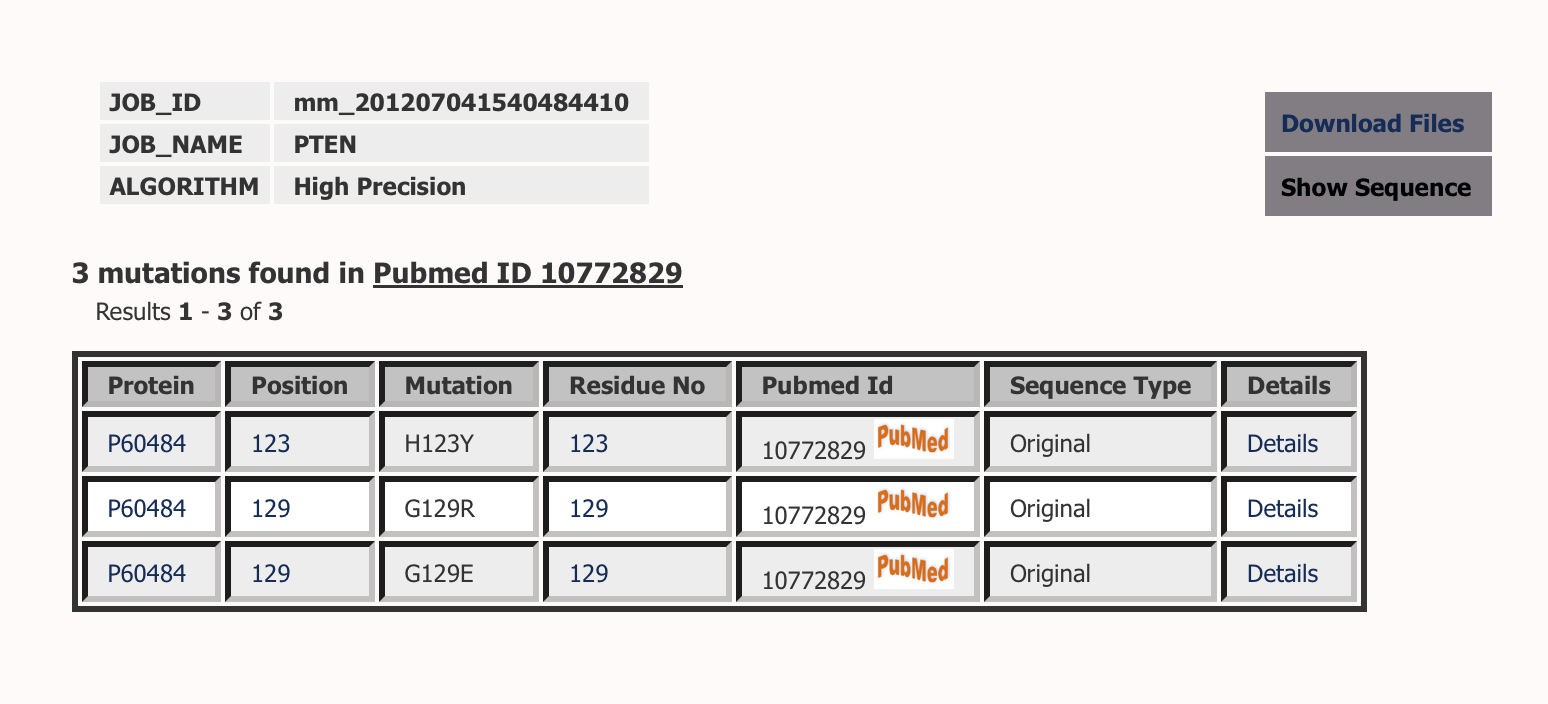
Gives you information about the mutation, protein sequence it was reported and the reference to the paper. Protein name is linked to Uniprot Server for more information. It displays the text with mention of mutation. Mutations are retrieved by automated pipeline so they are not validated. You can click on the PubMed Id to scan through the abstract to confirm it
We show some of the examples below used to evaluate the performance of MutationMapper. Some of the examples will show how the use of two different algorithm may be useful. It will also display the use of expressions will be helpful.
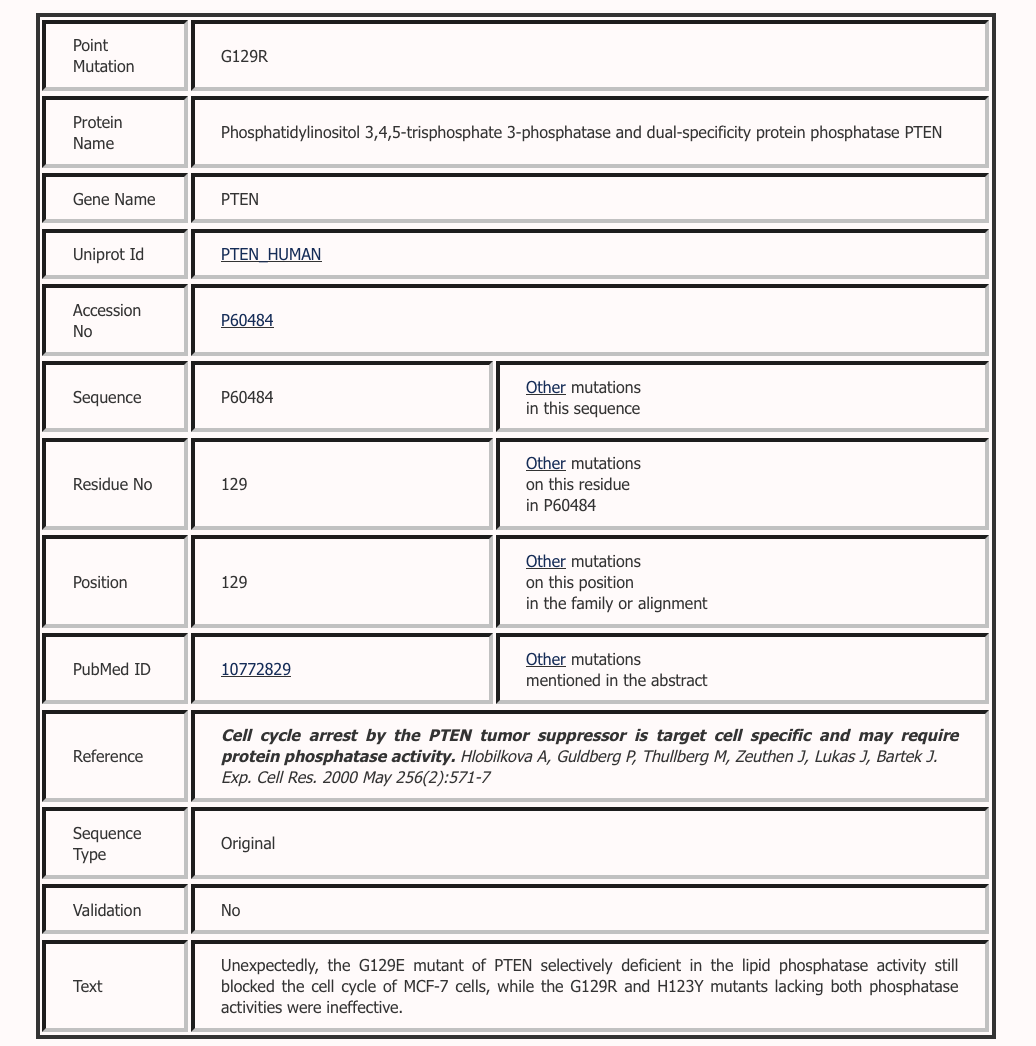
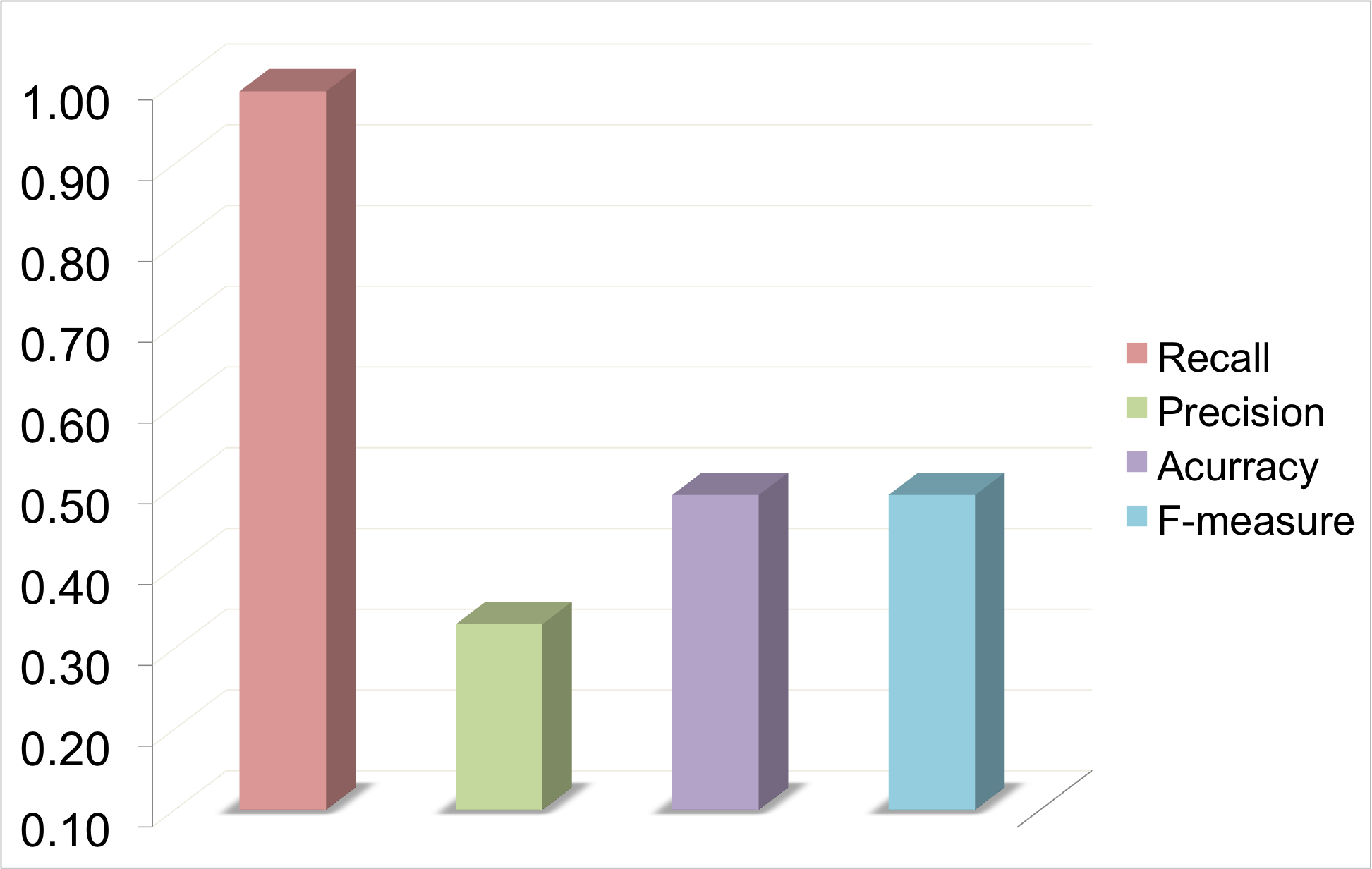
In this example, Uniprot Id was provided as the Input. Protein expressions were extarcted from Uniprot as shown below. These keywords were used to retrieve abstracts from PubMed. 6349 abstracts were retrieved and scanned for mutations. 454 mutations were reported in 311 abstracts. These muatations were mapped on to the protein sequence. It is one of the best examples where we have full recall and high precision. Hence, the accuracy and F-measure are good.

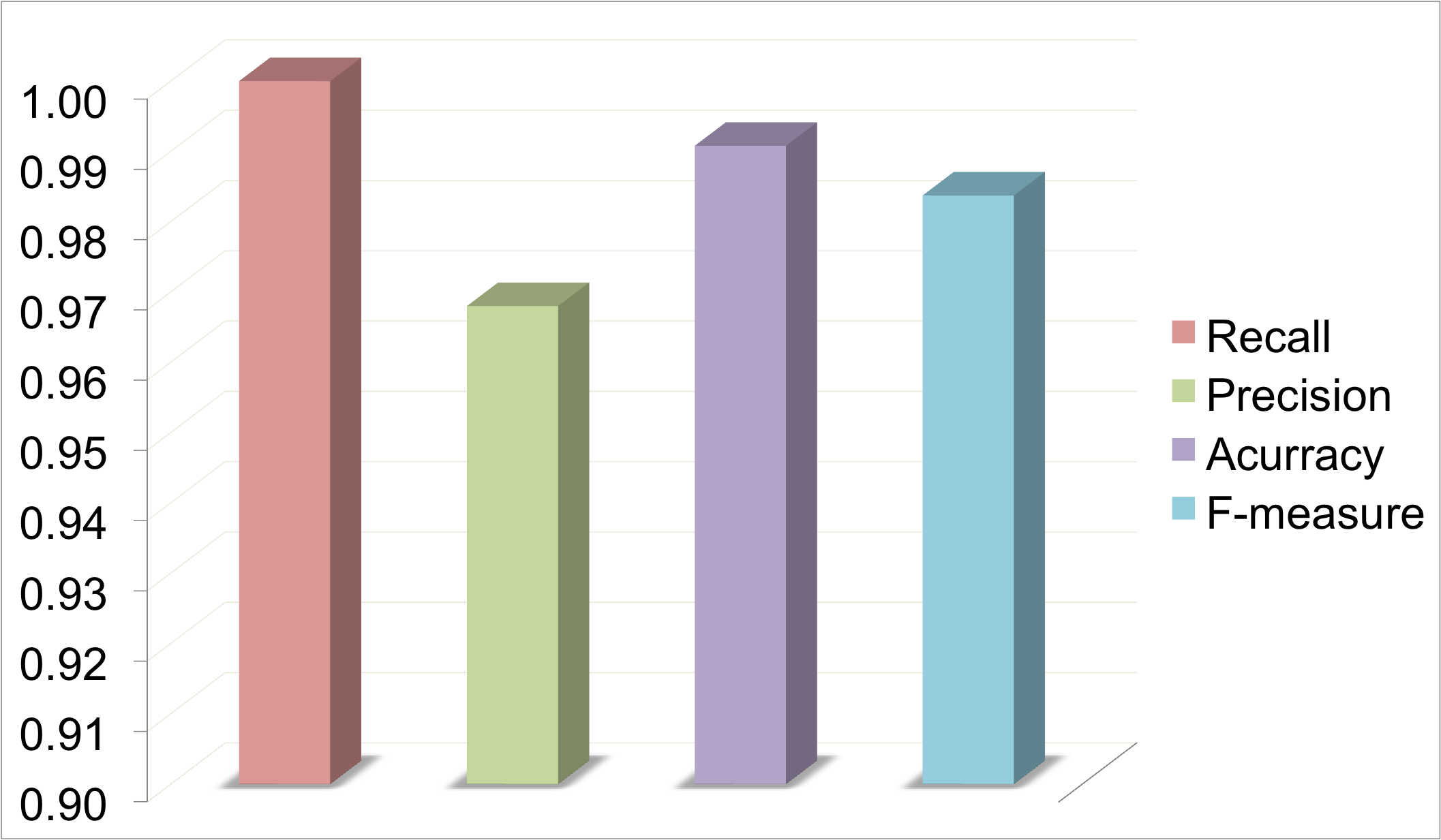
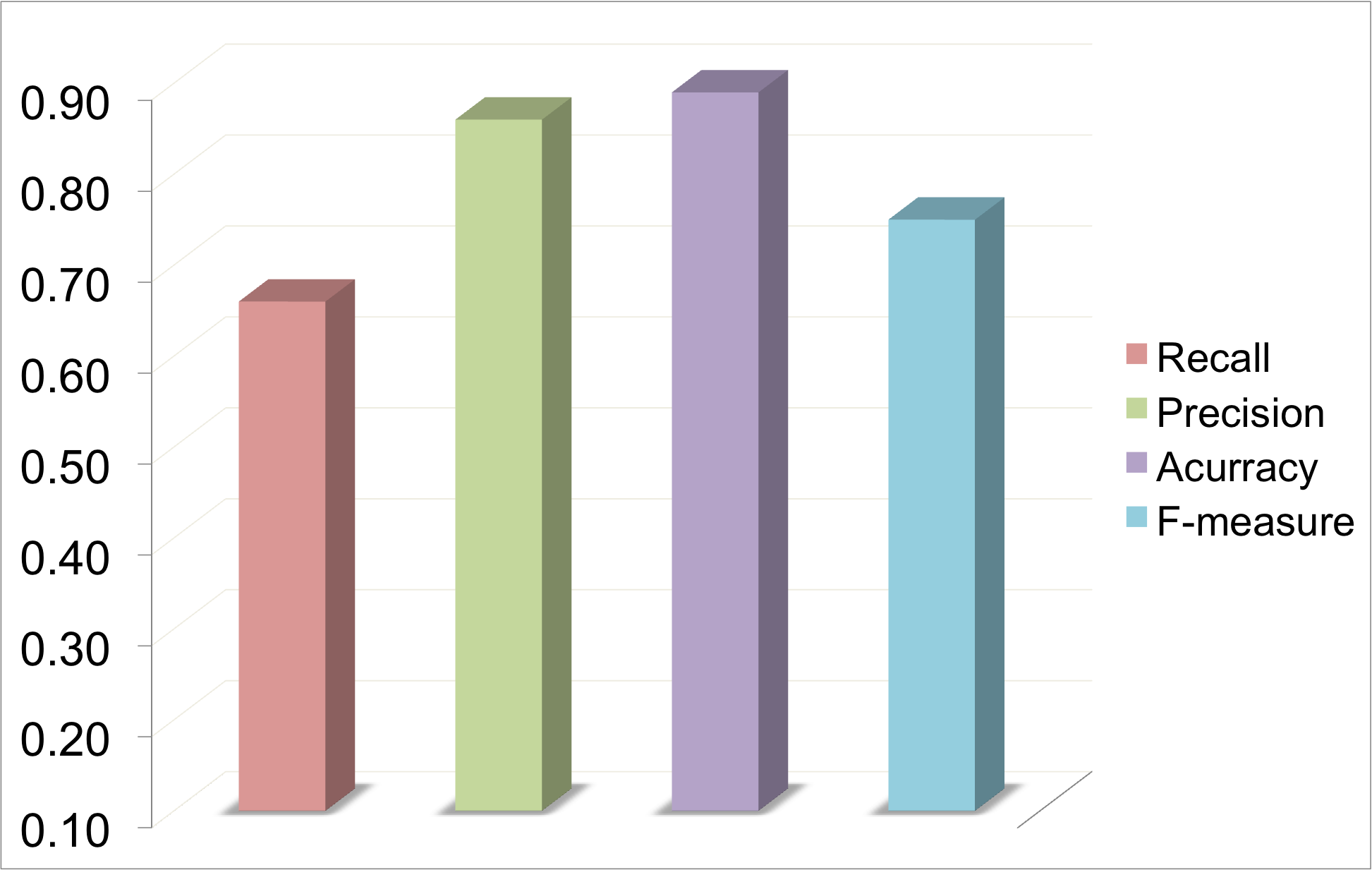
In this case, input was a multiple sequence alignmnet of Ionotropic glutamate receptor. 45339 abstracts were scanned for mutations and 1239 mutations were reported in 650 abstracts. In graphs below, there is a high precision in one case and high recall in the other.
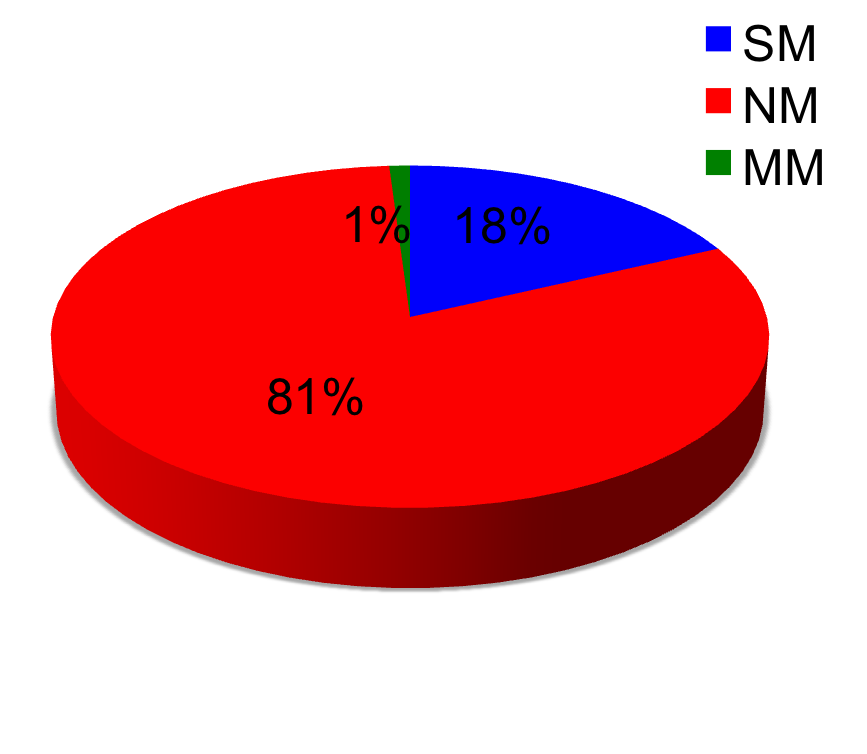
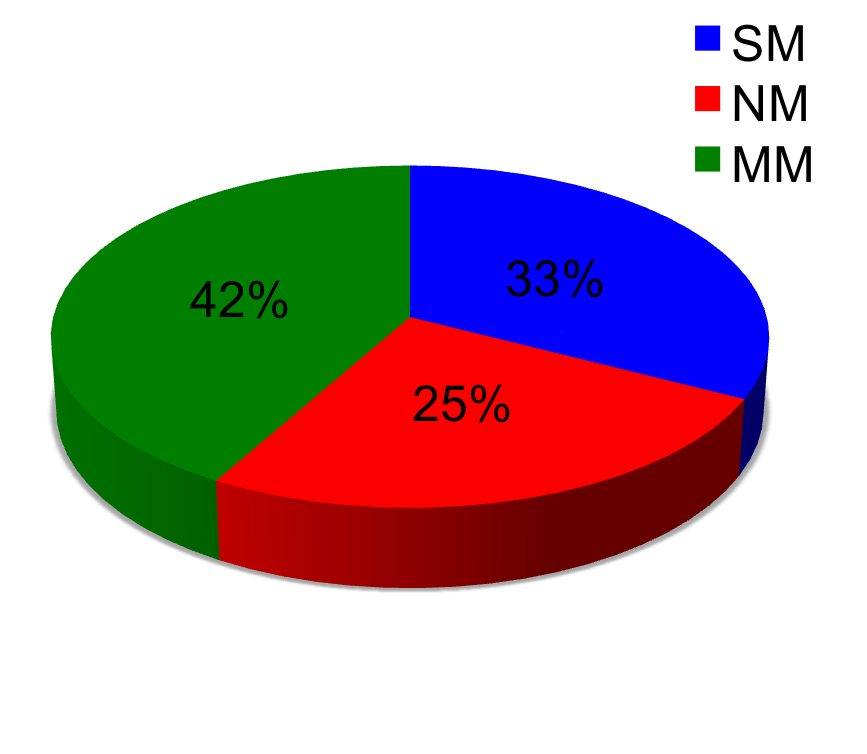
Pie-chart below shows the number of mutations mapped once (SM), number of mutations mapped more than once (MM), and number of mutations not mapped (NM). In case of high precision, the number of multi-mapped (MM) is very low (no false positives) but the number of non-mapped (NM) is high (increase in false negatives). In case of high recall, the number of single-mapped increases (mostly true positives) number of multi-mapped (MM)increases (increase in false positives), the non-mapped (NM) decreases (false negatives decreases). Most of the non-mapped are true negatives as they have not been reported on the protein of interest.
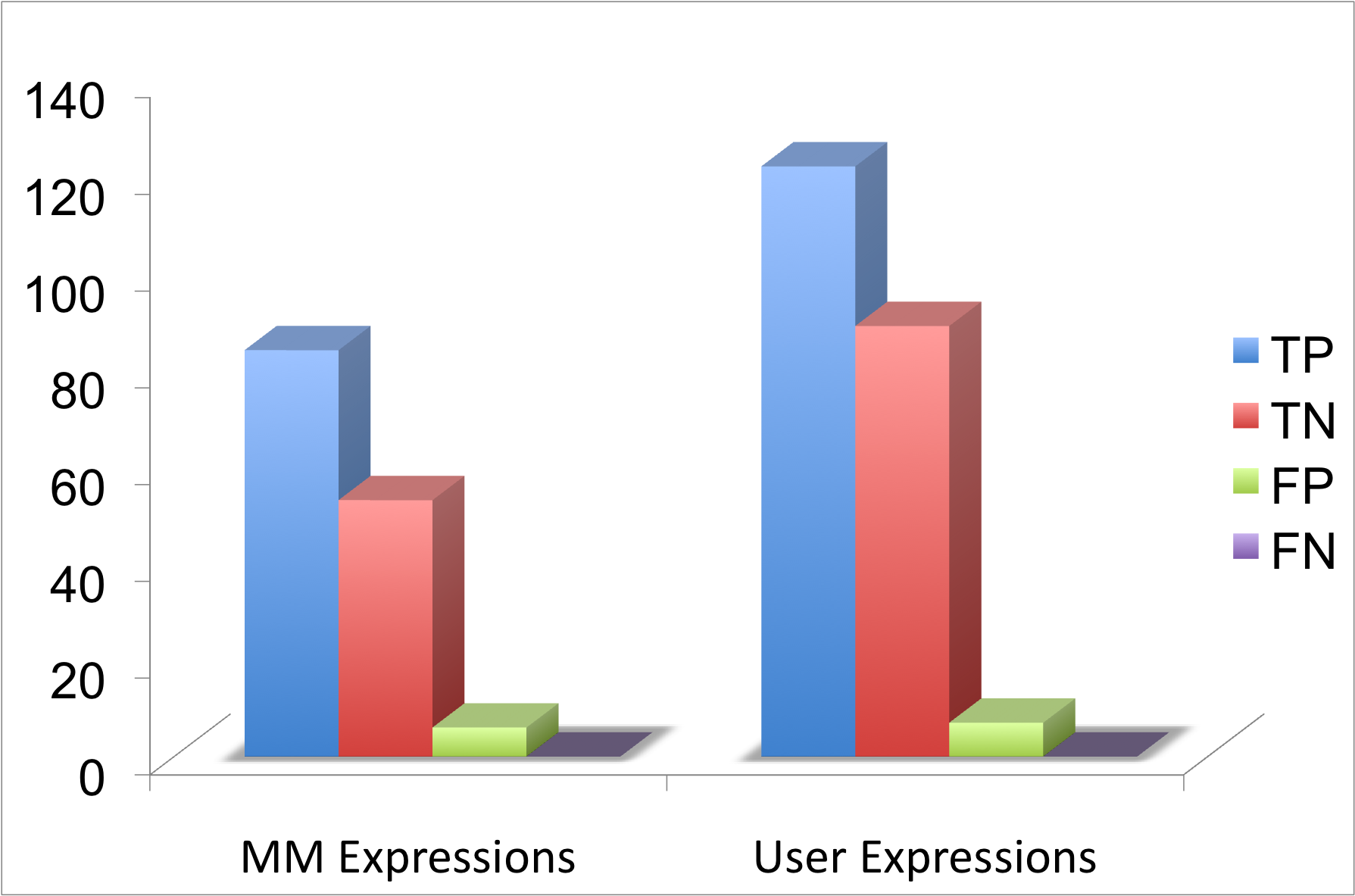
True positives are the mutations mapped on the correct sequence.
True negatives are the mutations not mapped as they are not reported on the sequence under consideration.
False Positives are the mutations mapped on an incorrect sequence .
False negatives are the mutations not mapped as the algorithm fails to map it on the correct sequence .
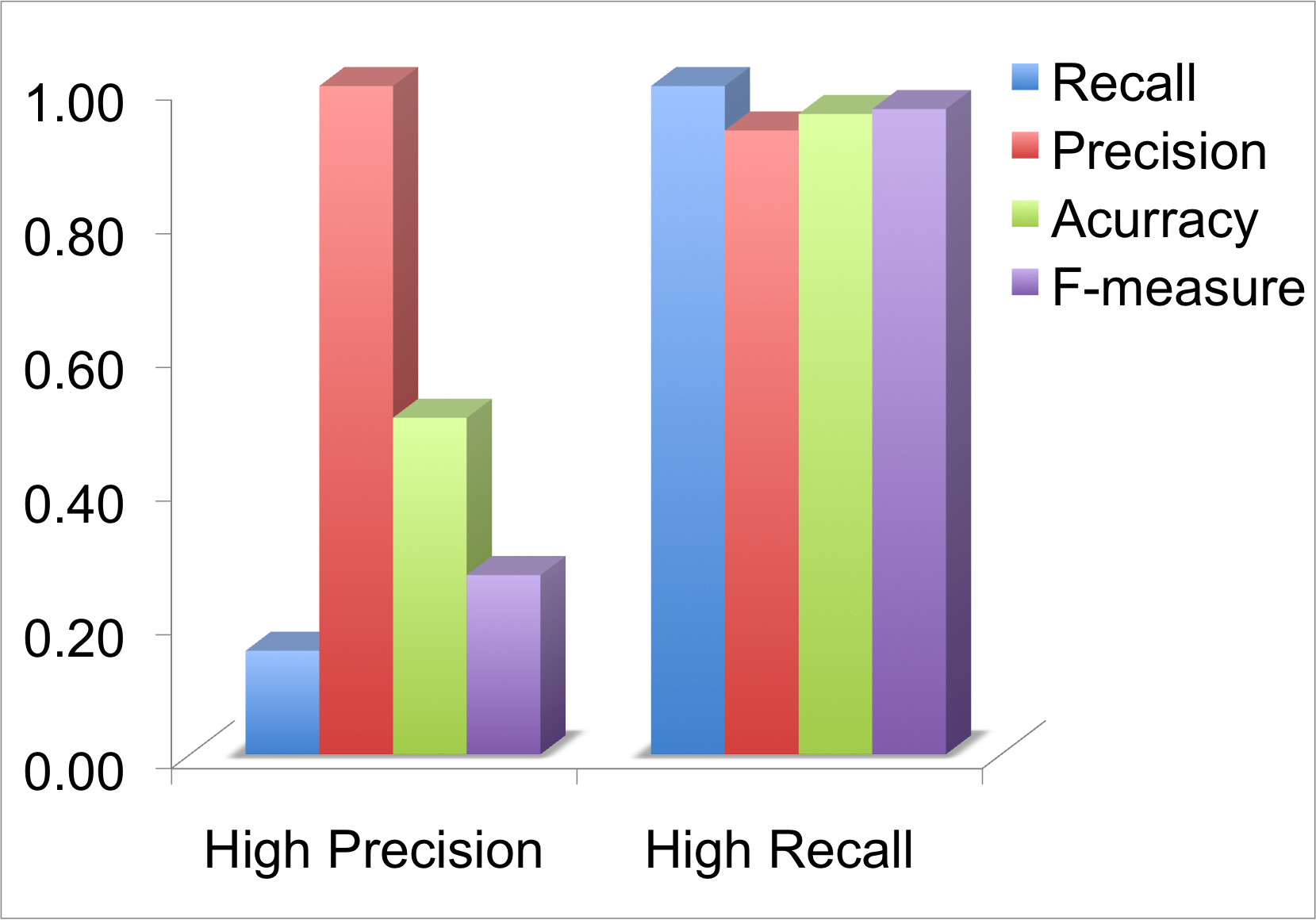
Two different algorithms are used to map mutations on to the protein sequence. They may give different results and be useful in some cases. In PTEN_HUMAN, the results generated by both the algorithms are same. But in examples like GRK1_RAT, GRK2_RAT and Ionotropic Glutamate Receptor (MSA) the use of two different algorithms gave different results.
Gives a high rate of true positives but may increase the number of false negatives. Hence, the recall may be low. In GRK1_RAT, precision is 100 precent but recall only 20 precent and there are many mutations which have not been mapped. In Ionotropic Glutamate Receptor (MSA), this algorithm gives a recall of 70 percent and precision of 90 percent.
Maps most of the mutations reported but may have a high rate of false positives and hence the precision decreases. In multiple sequence alignment there is usually an increase in number of multi-mapped mutations. In Ionotropic Glutamate Receptor (MSA), it is obvious that the use of this algorithm, has nearly full recall but the precision is low and the number of multi-mapped is high. In GRK1, both recall and precision are high.This indicates that High Recall does not necessarily always gives a low precision.
The examples above clearly show the difference in the results of the two algorithms. Results of High Precision has less noise in data as less false postives, but there is a chance of mutations not being reported. But in case of high recall, the noise in the data is high as the number of false positives can be high but there is a less chance of missing mutations reported. So it would be useful to browse the results of both the algorithms and may be one algorithm may provide better results than the other.
User has the option of input protein name expressions. This is useful when the Uniprot does not include the recommended names found in published text. When a user runs a job, they can download "SeqRegex.txt" file which lists the expressions used for the job. You can modify the file to include the most commonly used names and rerun the job with new set of expressions.
In case of NMDZ1_RAT, NR1 is the most common name used to report mutations in this protein. So adding NR1 to the input expression increases the hit rate and also improves the retrieval of abstract. 45 percent more abstracts were retrieved and there was 40 percent increase in the true positives.
"Matrix protein 2","BM2","M" |
P03369 "Gag-Pol polyprotein","Matrix protein p17","MA","Capsid protein p24","CA", "Spacer peptide p2","Nucleocapsid protein p7","NC","Transframe peptide","TF","p6-pol", "p6*","Protease","Reverse transcriptase/ribonuclease H","p51 RT","p15","Integrase", "IN","Pr160Gag-Pol","PR","Retropepsin","Exoribonuclease H","p66 RT","gag-pol" |
In some protein sequences, protein expressions downloaded from Uniprot may not be exhaustive or may include commonly occurring expressions. This effects the performance of MutationMapper and may not give the expected results. Some of the examples below shows how we have tried to address these issues.
In viral protein BM2_INBMP, the protein gene name is "M" and the recommended names is "BM2". In this case server retrieves too many abstracts and most of them are not relevant. Hence the algorithm ignores such expressions with one or two letters.